

重塑慢阻肺病管理格局,IL-33靶点迈向“上游阻断”精准新时代


南国风 暖,珠江潮涌。2026年5月末,呼吸领域迎来两场高水平学术盛会——2026慢阻肺国际学术会议与2026南山呼吸健康论坛在羊城广州接连举行。广州医科大学附属第一医院关伟杰教授和暨南大学附属第一医院刘升明教授分别于2026慢阻肺国际学术会议慢阻肺的发病机制分会场和2026南山呼吸健康论坛慢阻肺分论坛,围绕IL-33靶点在慢阻肺病中的潜力与未来前景进行了专题报告。报告聚焦慢阻肺病前沿,深度解读我国慢阻肺病疾病负担及IL-33核心病理机制,为慢阻肺病精准诊疗发展擘画全新方向。现将精彩内容整理如下,以飨读者。
暖,珠江潮涌。2026年5月末,呼吸领域迎来两场高水平学术盛会——2026慢阻肺国际学术会议与2026南山呼吸健康论坛在羊城广州接连举行。广州医科大学附属第一医院关伟杰教授和暨南大学附属第一医院刘升明教授分别于2026慢阻肺国际学术会议慢阻肺的发病机制分会场和2026南山呼吸健康论坛慢阻肺分论坛,围绕IL-33靶点在慢阻肺病中的潜力与未来前景进行了专题报告。报告聚焦慢阻肺病前沿,深度解读我国慢阻肺病疾病负担及IL-33核心病理机制,为慢阻肺病精准诊疗发展擘画全新方向。现将精彩内容整理如下,以飨读者。


2026南山呼吸健康论坛慢阻肺分论坛
冰山之下:我国慢阻肺病疾病负担沉重,临床治疗面临多重挑战
我国慢阻肺病患者人数庞大,有近1亿慢阻肺病患者,疾病负担沉重,成为我国第三大死亡原因1。回顾性队列研究SIRIUS I研究显示,即使规范使用吸入三联治疗,一年内仍有69%的患者出现≥1次中重度急性加重,25%的患者出现≥1次重度急性加重2。中国真实世界回顾性队列研究显示,接受三联治疗后患者仍面临较高的住院及死亡风险3。
究其原因,炎症是驱动慢阻肺病发生发展的核心病理机制,而黏液高分泌则为慢阻肺病的主要病理表现之一4。值得注意的是,目前的三联吸入治疗机制仍存在部分局限:在抗炎方面,吸入性糖皮质激素(ICS)仅抑制TNF-α/IL-6/IL-8促炎通路,较为局限,导致部分患者为ICS无应答者;而长效抗胆碱药物(LABA)/长效β2受体激动剂(LAMA)为长效支气管扩张剂,LABA主要扩张气管改善咳嗽清除、而LAMA则主要阻断黏液分泌反射来缓解症状,无直接抗炎效果,无法从根源上解决黏液高分泌问题5,6。研究表明,慢阻肺病患者接受吸入治疗后,仍78%的患者残留黏液栓,接受三联吸入治疗后超敏C反应蛋白(hs-CRP)、FeNO水平虽有下降,但仍残留炎症,嗜酸细胞计数在稳定期明显上升5,7。
基于此,精准靶向治疗成为主流研究方向,然而目前上市的生物制剂仅对GOLD指南推荐的嗜酸性粒细胞(EOS)计数>300/μL的炎症表型有效,且驱动慢阻肺病的炎症以中性粒细胞型和混合型为主,单纯EOS型患者较少8,9。此外,慢阻肺病患者慢性支气管炎、脓性黏液、黏液栓比例高,现有生物制剂尚缺乏针对黏液的相关证据,仍无法满足所有慢阻肺病患者的治疗需求5,10。因此,临床上亟需覆盖不同表型、兼顾复杂炎症控制与黏液管理的新型靶向药物。
曙光重现:IL-33——慢阻肺病治疗的破局新方向
气道炎症贯穿慢阻肺病的发病全程,是驱动肺功能进行性下降和慢阻肺病进展的关键因素11。气道上皮细胞受到外界刺激(如吸烟、过敏原、病毒、空气污染等)时主动释放免疫激活因子——上皮警报素(TSLP、IL-33、IL-25),是炎症反应的启动信号。上皮警报素尤其是TSLP、IL-33等可导致下游炎症细胞激活,生成IL-4、IL-5、IL-13等细胞因子,并促进嗜酸性粒细胞的浸润,驱动症状加重和疾病进展12。因此,靶向上游IL-33/TSLP上皮警报素正逐渐成为呼吸系统疾病治疗的前沿方向。
IL-33作为关键的上游警报素之一,在慢阻肺病中与炎症反应和黏液高分泌密切相关。IL-33特异性受体ST2广泛分布于上皮细胞、内皮细胞和免疫细胞表面,是激活慢阻肺病炎症通路的重要上游驱动因子。IL-33与ST2结合激活下游通路,介导气道慢性炎症、黏液高分泌及气道重塑,导致慢阻肺病发生发展与急性加重12,13。
对于慢阻肺病,IL-33可同步激活1/2/3型全炎症通路:
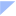
1)对于2型炎症,IL-33可激活ILC2细胞,促进Th2细胞分化,释放IL-4、IL-5、IL-13等炎症因子,促进EOS募集、成熟和存活并向气道迁移和浸润,继而激活巨噬细胞,共同导致炎症反应和气道重塑,此外IL-13与IL-4还会促进黏蛋白MUC5AC表达,诱导杯状细胞增生及黏液纤毛功能障碍,导致黏液高分泌和气道阻塞12,14-18。
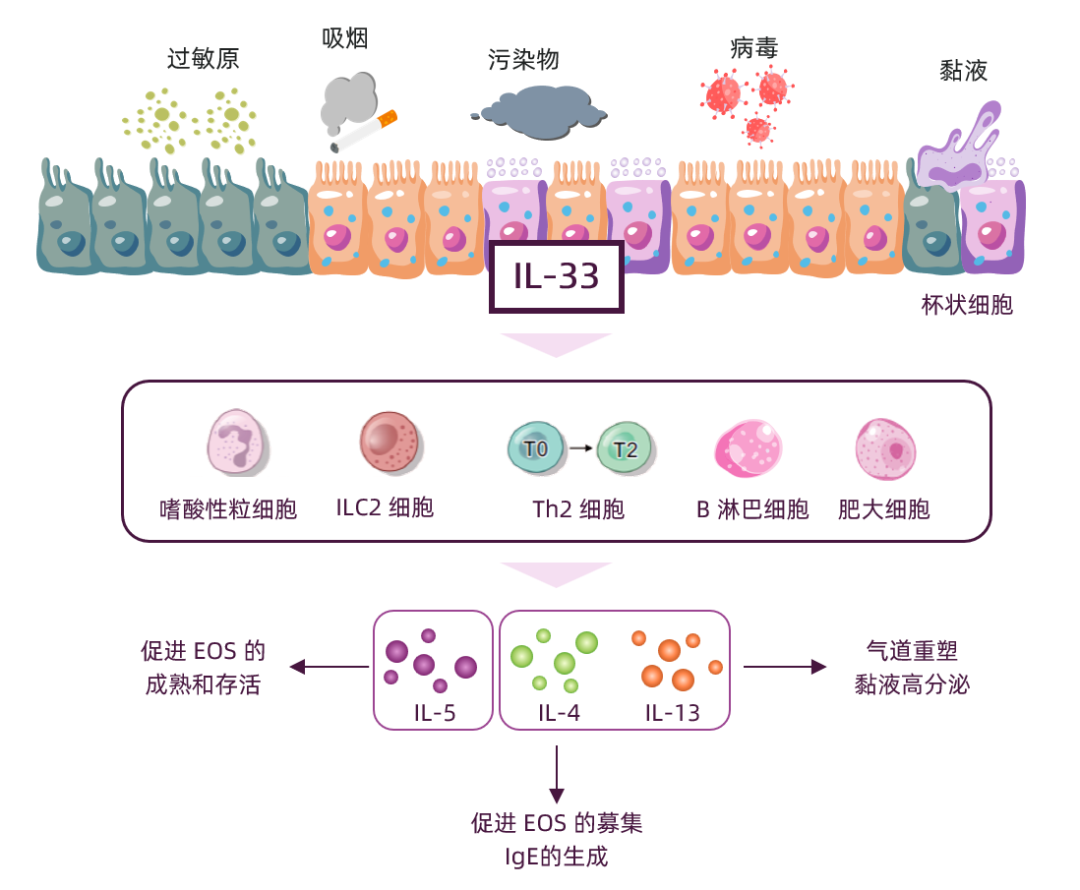
图1 IL-33激活2型炎症通路
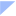
2)对于1/3型炎症,IL-33激活Th1、ILC3细胞等,促进IFN-γ、IL-17的释放。IFN-γ激活肺泡巨噬细胞,释放弹性蛋白酶和基质金属蛋白酶,导致肺气肿;增强CD8⁺ T细胞的细胞毒性,损伤气道上皮细胞。IL-17促进中性粒细胞的募集和活化,造成气道黏膜损伤,促进气道上皮杯状细胞增生与黏液高分泌,堵塞气道15,19-22。
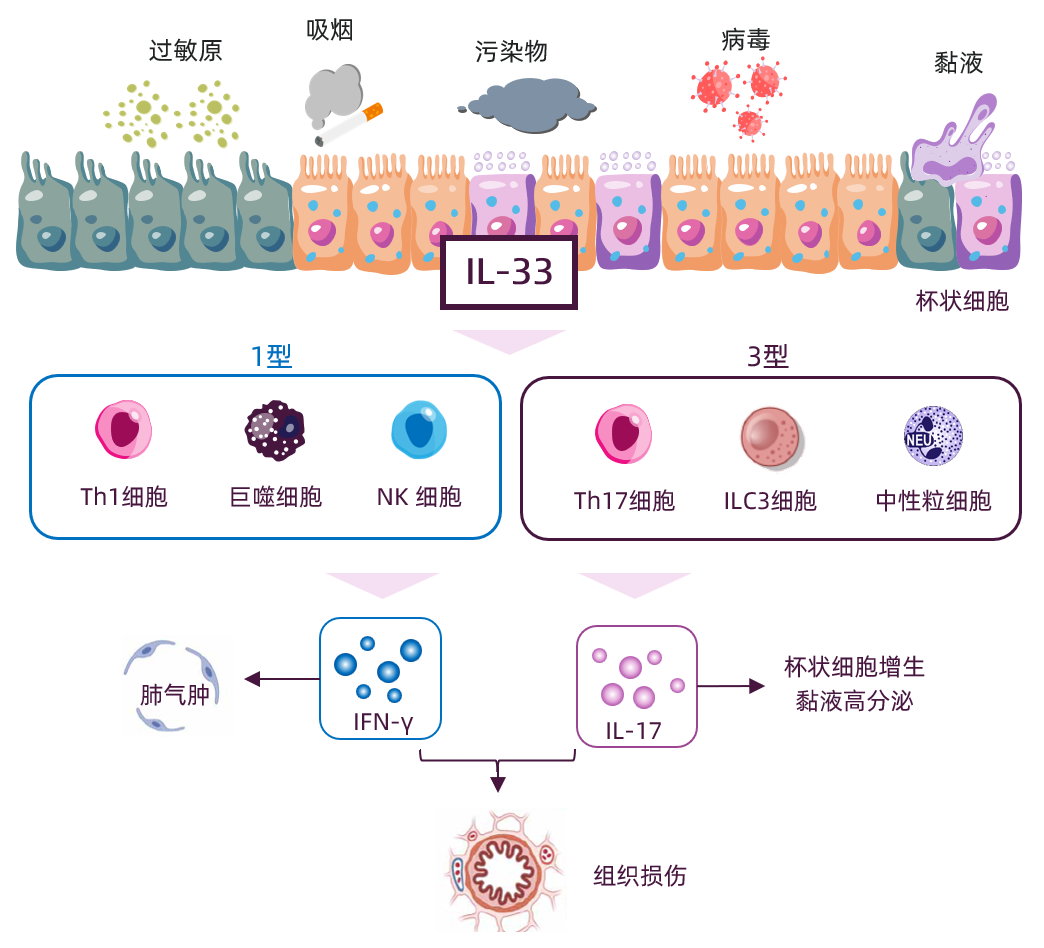
图2 IL-33激活1/3型炎症通路
此外,IL-33存在还原型和氧化型两种形态,分别通过不同机制驱动慢阻肺病病理进程:
IL-33初始释放时为还原型IL-33RED,储存于上皮细胞、内皮细胞等结构细胞的细胞核内,暴露于细胞外环境后发生构象转换,转化为氧化型IL-33OX23,24。
气道上皮细胞受损后,还原型IL-33RED通过与多种炎性细胞表面的ST2受体结合,促进下游炎症反应的发生,触发1/2/3型炎症反应,导致慢阻肺病症状加重,伴随急性加重风险升高及肺组织损伤。氧化型IL-33OX不与ST2受体结合,而是与可调节上皮稳态的RAGE/EGFR形成复合物,破坏气道稳态,增加黏液分泌导致杯状细胞增生、黏液高分泌/黏液栓形成,最终引发气道屏障破坏与结构重塑。两条通路共同作用,最终引发慢阻肺病以下特征性病理改变与临床表现:1)肺气肿、呼吸困难与慢性咳嗽;2)黏液高分泌;3)慢性炎症(促炎信号传导与急性加重);4)小气道阻塞及肺实质破坏;5)纤维化伴上皮重塑与损伤25。
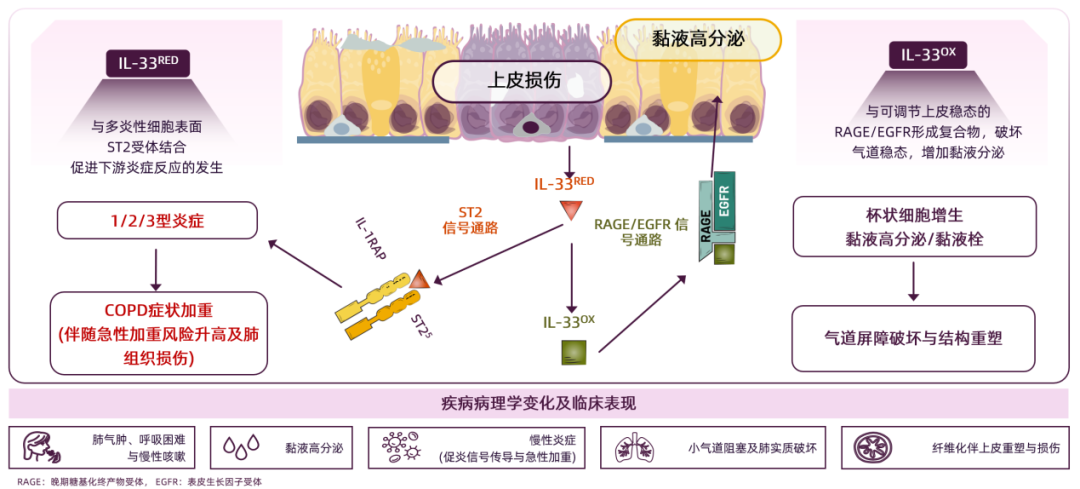
图3 还原型IL-33RED和氧化型IL-33OX驱动慢阻肺病相关病理进程的分子机制
综上所述,IL-33通过同步激活1/2/3型全炎症通路,驱动气道慢性炎症、肺气肿、黏液高分泌等多种病理改变;其还原型IL-33RED和氧化型IL-33OX两种形态通过介导炎症反应和形成黏液高分泌/黏液栓,共同促进慢阻肺病的发生发展与急性加重。因此,IL-33成为慢阻肺病治疗领域极具潜力的上游靶点之一。
小结
慢阻肺病作为我国居民健康的重大威胁,虽然目前吸入治疗已被广泛应用,但仍有大量患者在规范吸入的前提下饱受反复急性加重、肺功能持续下降和黏液栓残留的困扰。究其原因,慢阻肺病具有异质性强且病程长、炎症复杂且伴有黏液高分泌的特征。在这一背景下,上游警报素IL-33靶点为慢阻肺病治疗开辟了全新的路径。
未来,随着IL-33靶点研究的不断深入探索,有望推动我国慢阻肺病诊疗水平迈向新高度,助力实现GOLD 2026提出的“无急性加重”低疾病活动状态的核心管理目标,开启慢阻肺病精准靶向治疗的新时代。
﹀
﹀
﹀
审批编号:CN-185334 到期时间:2027-06-05
本材料由阿斯利康提供,仅供医疗卫生专业人士进行医学科学交流,不用于推广目的

