

Cell Death & Disease | 复旦中山医院团队揭示软骨细胞PKM2二聚化通过ERK-MFN1轴破坏线粒体稳态驱动骨关节炎
Metadata Card
文献题目:Dimeric PKM2 in chondrocytes impairs mitochondrial homeostasis in osteoarthritis
发表期刊:Cell Death & Disease
发表时间:26.3.25
影响因子:9.6(中科院1区)
通讯作者/单位: Chi Zhang @ 复旦大学附属中山医院
一句话总结:研究发现PKM2从四聚体向二聚体的构象转换通过激活ERK信号通路抑制MFN1,导致线粒体功能障碍并破坏软骨细胞外基质稳态。
Graphical Abstract
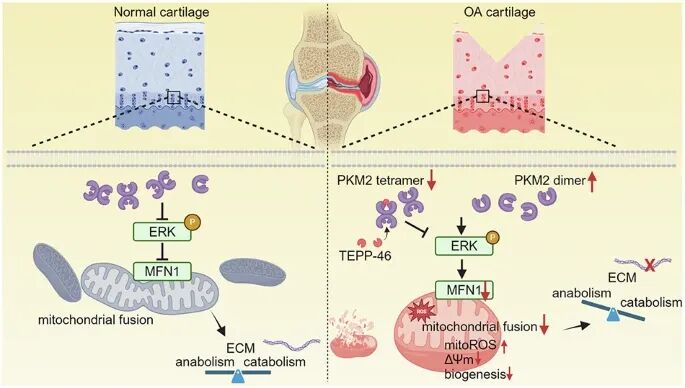
Abstract
中文摘要:
软骨降解被认为是终末期骨关节炎 (OA)的标志,其特征在于细胞外基质(ECM)的显著改变。本研究检测了丙酮酸激酶肌肉2型(PKM2)二聚化在骨关节炎软骨降解和ECM稳态中的作用。生物信息学分析确定了OA软骨中PKM的上调,特别是在纤维软骨亚群中。在人和鼠OA软骨中均观察到PKM2的升高表达和二聚化。在内侧半月板(DMM)不稳定的鼠OA模型中,软骨细胞特异性PKM2缺乏以及使用TEPP-46(一种PKM2四聚体稳定剂)治疗可减少OA进展并促进软骨基质产生。从机制上讲,PKM2缺乏或四聚体稳定通过破坏PKM2-ERK相互作用促进线粒体融合并保留线粒体功能,导致ERK依赖性线粒体融合蛋白1(MFN1)上调,而不是线粒体融合蛋白2(MFN2)上调。值得注意的是,AAV介导的MFN1敲除消除了PKM2缺乏的软骨保护作用。这些发现表明,靶向PKM2二聚化可能是缓解OA的一种有前途的治疗策略。
(OA)的标志,其特征在于细胞外基质(ECM)的显著改变。本研究检测了丙酮酸激酶肌肉2型(PKM2)二聚化在骨关节炎软骨降解和ECM稳态中的作用。生物信息学分析确定了OA软骨中PKM的上调,特别是在纤维软骨亚群中。在人和鼠OA软骨中均观察到PKM2的升高表达和二聚化。在内侧半月板(DMM)不稳定的鼠OA模型中,软骨细胞特异性PKM2缺乏以及使用TEPP-46(一种PKM2四聚体稳定剂)治疗可减少OA进展并促进软骨基质产生。从机制上讲,PKM2缺乏或四聚体稳定通过破坏PKM2-ERK相互作用促进线粒体融合并保留线粒体功能,导致ERK依赖性线粒体融合蛋白1(MFN1)上调,而不是线粒体融合蛋白2(MFN2)上调。值得注意的是,AAV介导的MFN1敲除消除了PKM2缺乏的软骨保护作用。这些发现表明,靶向PKM2二聚化可能是缓解OA的一种有前途的治疗策略。
英文摘要:
Cartilage degradation is considered a hallmark of end-stage osteoarthritis (OA), characterized by significant alterations in the extracellular matrix (ECM). This study examines the role of pyruvate kinase muscle type 2 (PKM2) dimerization in cartilage degradation and ECM homeostasis in OA. Bioinformatic analyses identified an upregulation of PKM in OA cartilage, particularly within fibrocartilage subpopulations. Elevated expression and dimerization of PKM2 were observed in both human and murine OA cartilage. Chondrocyte-specific PKM2 deficiency, along with treatment using TEPP-46, a PKM2 tetramer stabilizer, reduced OA progression and promoted cartilage matrix production in a murine OA model with destabilization of the medial meniscus (DMM). Mechanistically, PKM2 deficiency or tetramer stabilization promoted mitochondrial fusion and preserved mitochondrial function via disruption of PKM2–ERK interaction, resulting in ERK-dependent upregulation of mitofusin 1 (MFN1), but not mitofusin 2 (MFN2). Notably, AAV-mediated MFN1 knockdown abrogated the chondroprotective effects of PKM2 deficiency. These findings indicate that targeting PKM2 dimerization may represent a promising therapeutic strategy for mitigating OA.
Background & Problem
背景:骨关节炎(OA)是一种严重的慢性关节疾病,其核心病理过程是软骨细胞外基质(ECM)的持续降解。由于软骨处于无血管、缺氧环境,软骨细胞高度依赖糖酵解获取能量,而丙酮酸激酶M2(PKM2)正是这一代谢途径的关键限速酶。
痛点:尽管已知PKM2在OA中表达升高,且与细胞衰老、炎症有关,但其独特的“构象切换”(二聚体vs.四聚体)如何具体影响线粒体动力学及软骨稳态,此前尚不清楚。
Methodology
策略:研究团队结合了生物信息学分析、临床标本验证、基因工程小鼠模型及药理学干预手段。
逻辑:首先通过单细胞测序(scRNA-seq)定位PKM在OA中的异常表达;随后利用siRNA和条件性敲除(Pkm2icKO)小鼠验证PKM2的功能;最后通过共免疫沉淀(Co-IP)和线粒体压力测试探讨PKM2、ERK与MFN1之间的分子级联反应。
Key Results & Interpretation
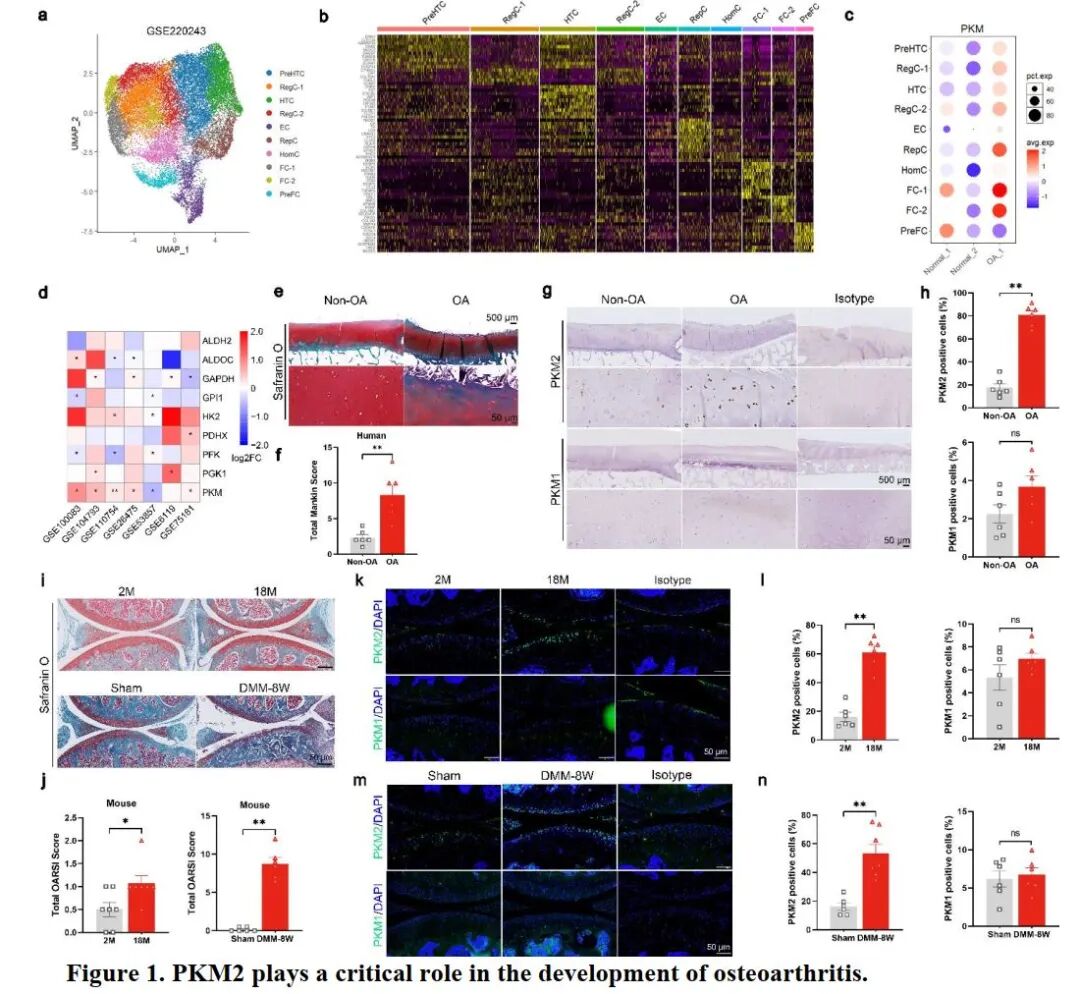
[A-C]:通过单细胞 RNA 测序 (scRNA-seq) 发现,在 10 个软骨细胞亚群中,PKM 在所有亚群中普遍上调,且在调控软骨纤维化的 FC-2 亚群中表达最高。
[D]:整合 7 个公共转录组数据集,证实 PKM 是 OA 软骨中唯一持续上调的糖酵解限速酶。
[E-F]:人类临床样本显示 OA 软骨中蛋白聚糖显著丢失,Mankin 评分大幅上升。
[G-H]:免疫组化证实 OA 软骨中 PKM2 蛋白显著升高,而 PKM1 水平在组间无明显差异。
[I-J]:在 DMM 诱导及自然衰老的小鼠模型中,SO&FG 染色及 OARSI 评分确认了软骨损伤的加重。
[K-N]:免疫荧光分析进一步证实小鼠 OA 软骨中 PKM2 表达激增,且伴随明显的核转位现象。
发现点2:下调 PKM2 表达可显著维持软骨基质代谢平衡,缓解软骨退变
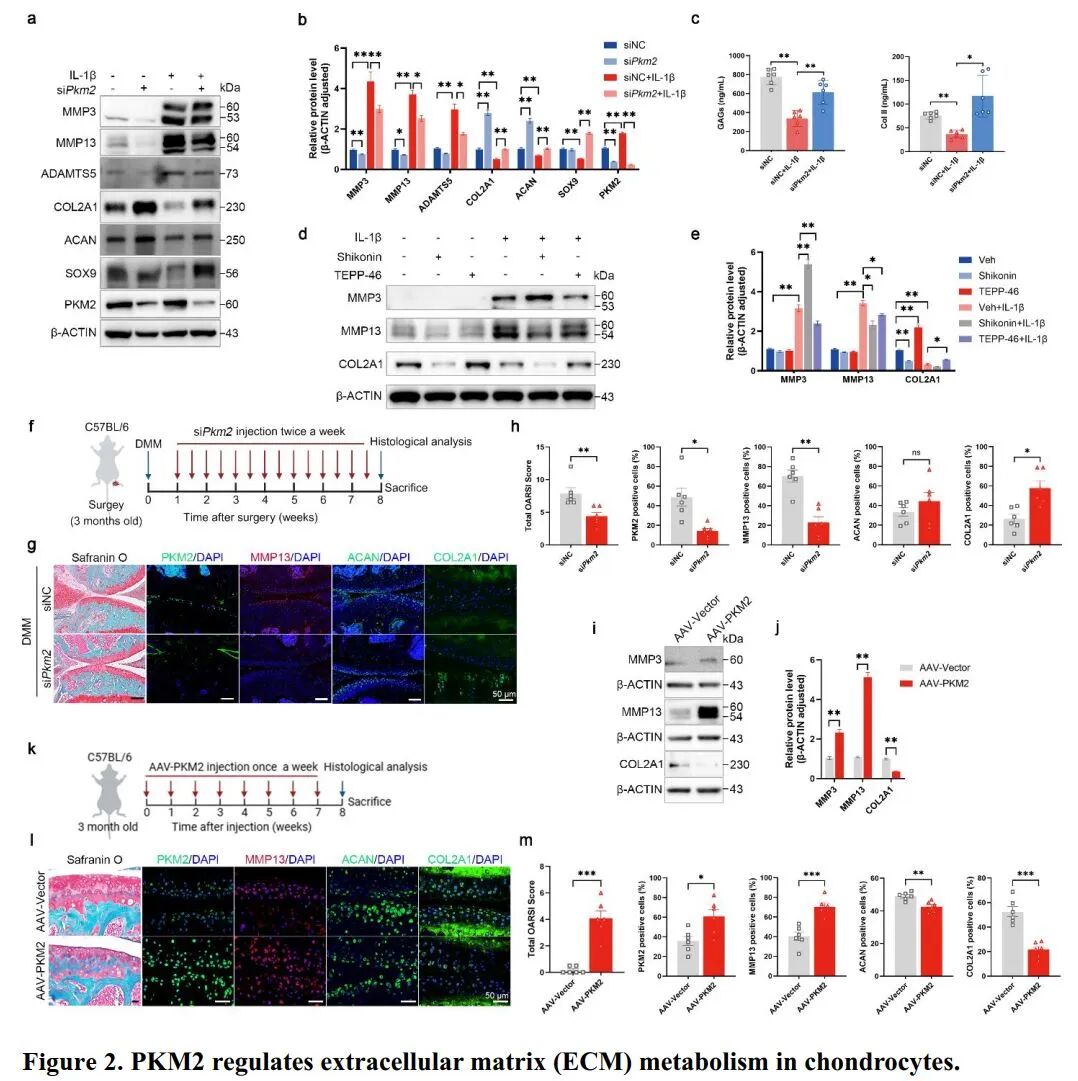
[A-B]:在 IL-1β 诱导的炎症模型中,siPkm2 显著降低了基质降解酶 (MMP3, MMP13, ADAMTS5) 的水平,并挽救了基质成分 (COL2A1, ACAN, SOX9) 的表达。
[C]:培养上清液检测显示,Pkm2 敲低显著提升了糖胺聚糖 (GAGs) 和 II 型胶原的含量。
[D-E]:药理学对比发现,四聚体稳定剂 TEPP-46 模拟了敲低效果,而抑制剂 Shikonin 则加剧了基质分解。
[F-H]:在 DMM 小鼠体内,关节内注射 siPkm2 显著减轻了软骨侵蚀,下调了 MMP13 并上调了 COL2A1。
[I-M]:利用 AAV 介导的 PKM2 过表达实验显示,PKM2 高表达直接诱发了自发性软骨损伤和基质代谢失衡。
发现点3:软骨细胞特异性敲除 PKM2 可显著缓解 DMM 诱导的 OA 进展及疼痛
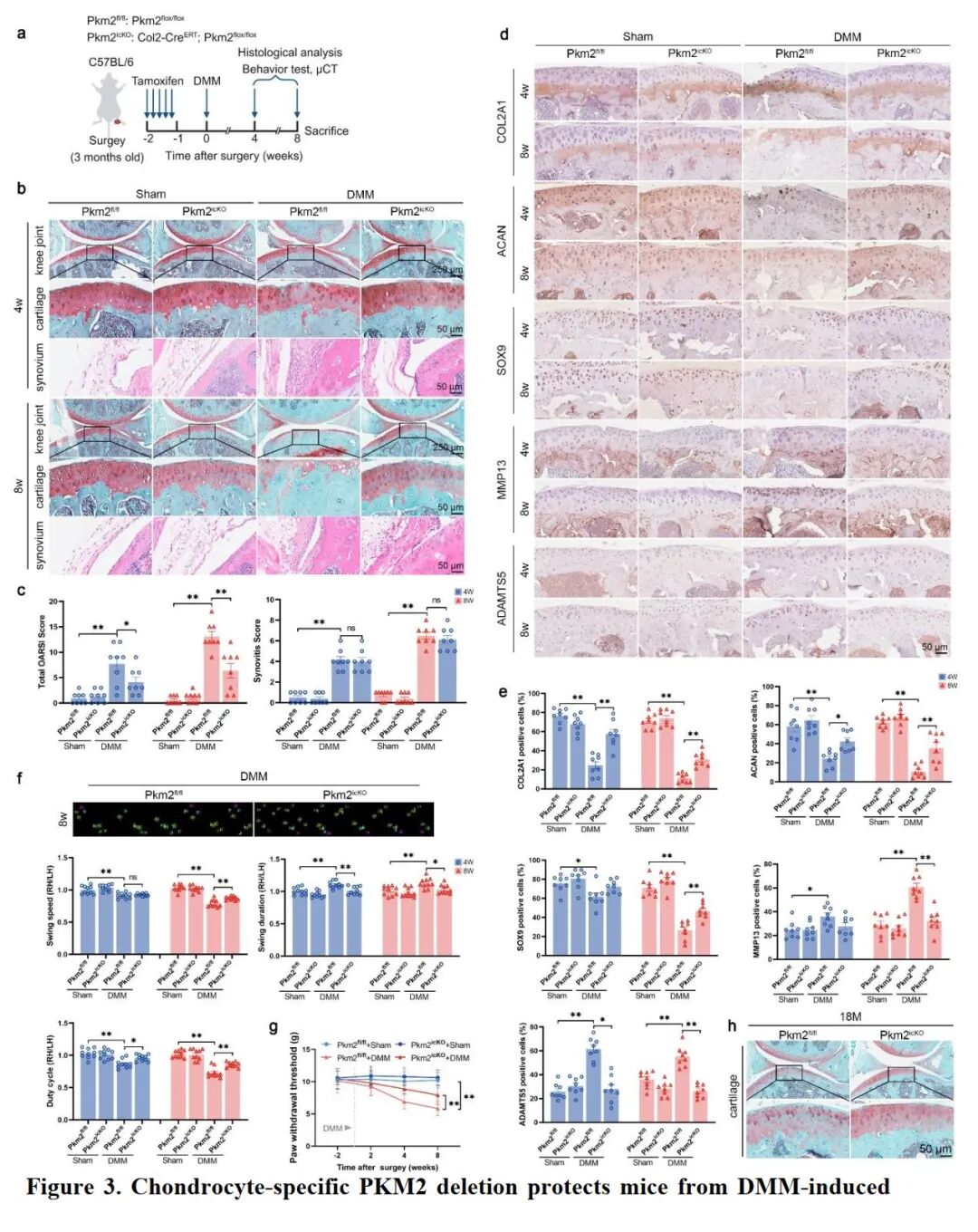
[A-C]:通过 tamoxifen 诱导构建 Pkm2icKO 小鼠,SO&FG 染色显示其在 DMM 手术 4 周和 8 周后均保留了更完整的软骨结构,OARSI 评分显著低于对照组。
[D-E]:免疫组化证实敲除组小鼠软骨中 COL2A1、ACAN 和 SOX9 水平更高,而 MMP13 和 ADAMTS5 的表达受到明显抑制。
[F]:CatWalk步态分析显示,Pkm2icKO 小鼠的摆动速度 (swing speed) 和步态周期 (duty cycle) 显著改善,说明运动功能得到保护。
[G]:Von Frey 测试结果显示敲除组小鼠具有更高的机械痛阈值,提示其疼痛症状得到有效缓解。
[H]:在 18 个月大的自然衰老小鼠中,Pkm2icKO 同样展现出更强的软骨完整性。
发现点4:PKM2 构象失衡(二聚体增加)是驱动 OA 病理改变的关键开关
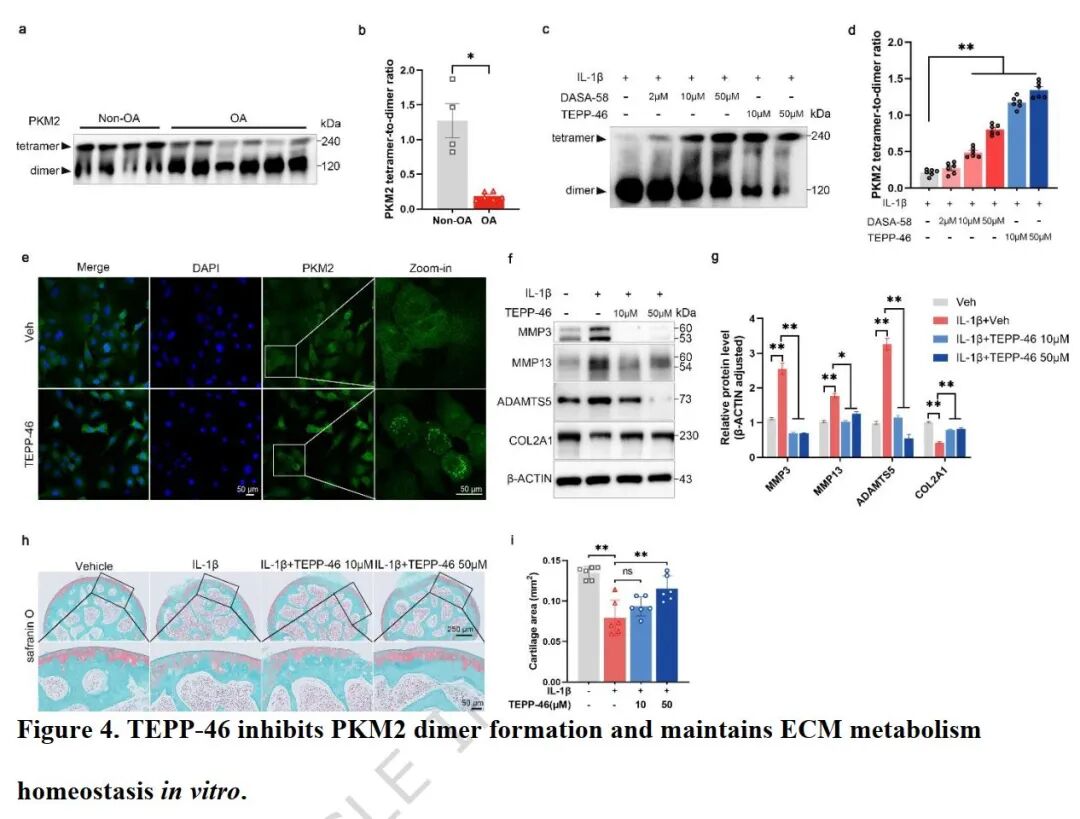
[A-B]:化学交联实验对比发现,人类 OA 软骨中的 PKM2 二聚体明显增加,而四聚体显著减少。
[C-D]:在 IL-1β 刺激下,四聚体稳定剂 (TEPP-46 或 DASA-58) 能以剂量依赖的方式减少二聚体形成。
[E]:免疫荧光显示 TEPP-46 促使 PKM2 在胞质和胞核中形成点状和聚集信号。
[F-G]:TEPP-46 治疗显著抑制了 IL-1$\beta$ 诱导的 MMPs 表达,并增强了 COL2A1 表达。
[H-I]:离体股骨头外植体实验证实,TEPP-46 能有效减轻炎症引起的软骨面积损失。
发现点5:药理学稳定 PKM2 四聚体能有效缓解软骨退变及下层骨重塑
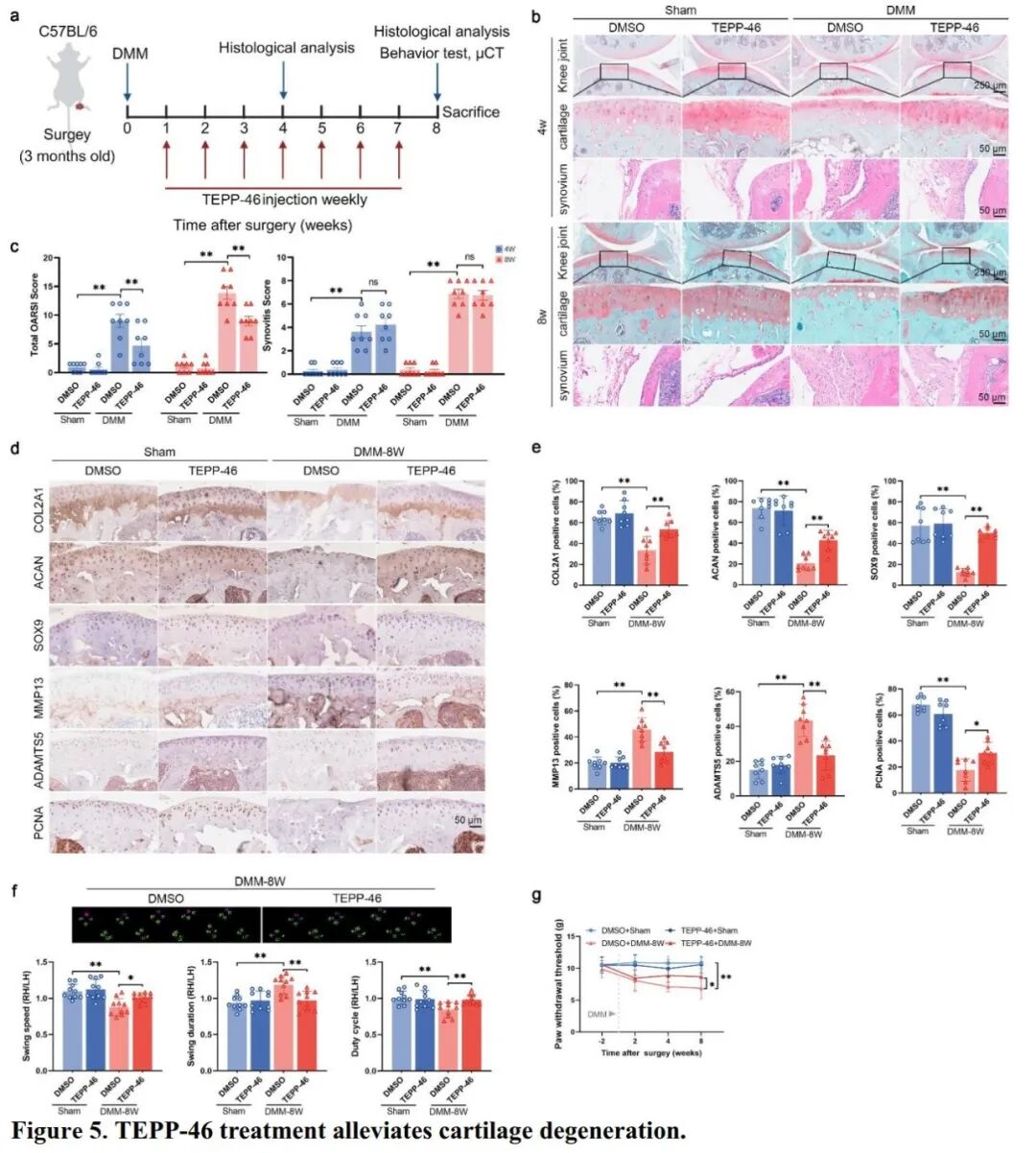
[A]:展示了 DMM 手术后每周进行 TEPP-46 关节内注射的实验设计。
[B-C]:组织学分析显示,TEPP-46 显著减少了 DMM 诱导的软骨侵蚀,降低了总 OARSI 评分,但对滑膜炎影响较小。
[D-E]:免疫组化结果确认,TEPP-46 增加了 COL2A1、ACAN 及 PCNA 的水平,并降低了降解酶的表达。
[F-G]:CatWalk 及 Von Frey 测试再次证实 TEPP-46 能够显著提升小鼠的机械痛阈值并改善步态参数。
发现点6:PKM2 缺失或四聚化通过保护线粒体稳态防止软骨细胞功能衰竭
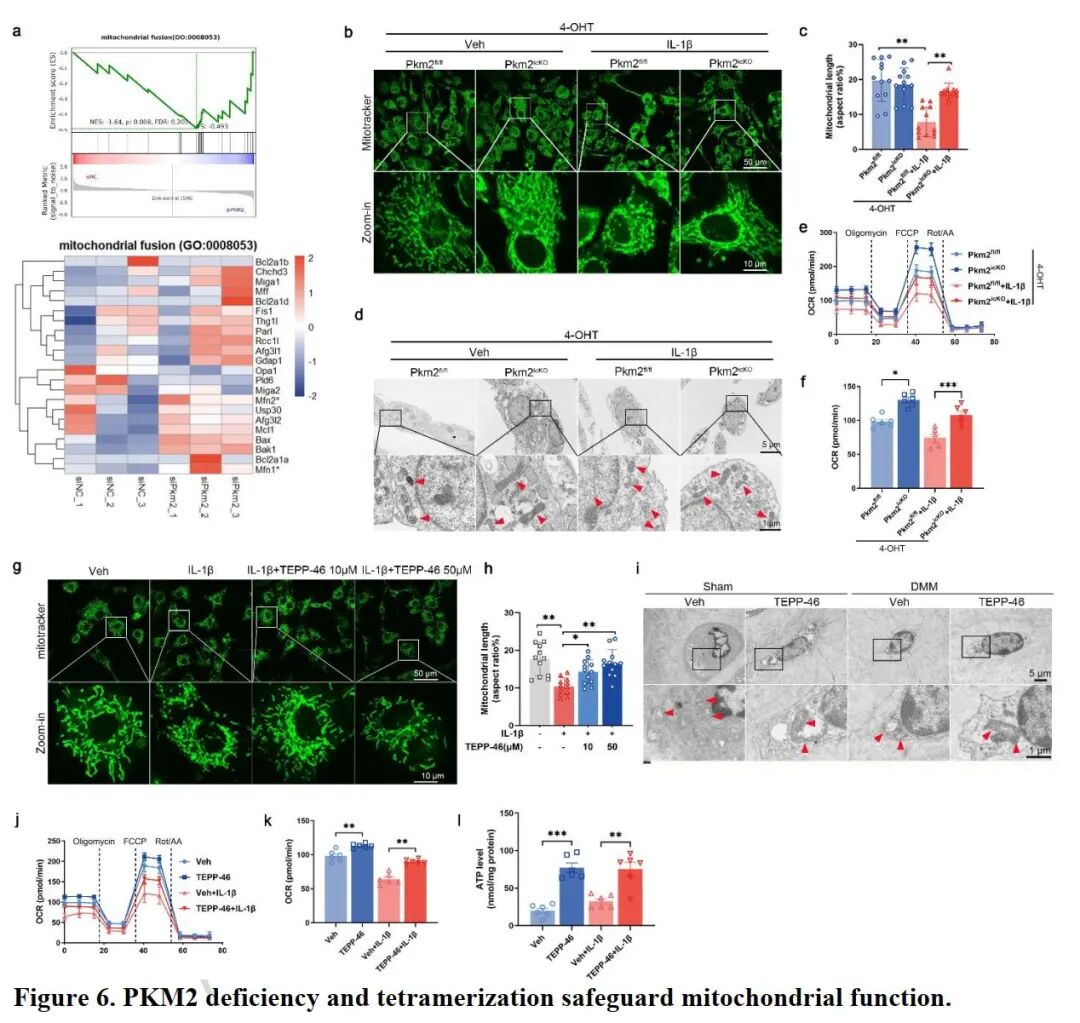
[A]:RNA-seq 富集分析表明 Pkm2 敲低显著激活了线粒体融合及氧化磷酸化通路。
[B-D]:MitoTracker 染色及透射电镜 (TEM) 显示,Pkm2 缺失可逆转 IL-1β 导致的线粒体碎片化,恢复长管状结构。
[E-F]:Seahorse 实验证实 Pkm2 缺失显著提升了细胞的基础耗氧率 (OCR)。
[G-H]:TEPP-46 同样在体外改善了线粒体形态,使其长宽比显著增加。
[I-L]:体内 TEM 及功能测试 (OCR, ATP) 表明,稳定四聚体能保护线粒体超微结构并提升 ATP 产量。
发现点7:MFN1(而非 MFN2)是介导 PKM2 缺失发挥软骨保护作用的关键分子
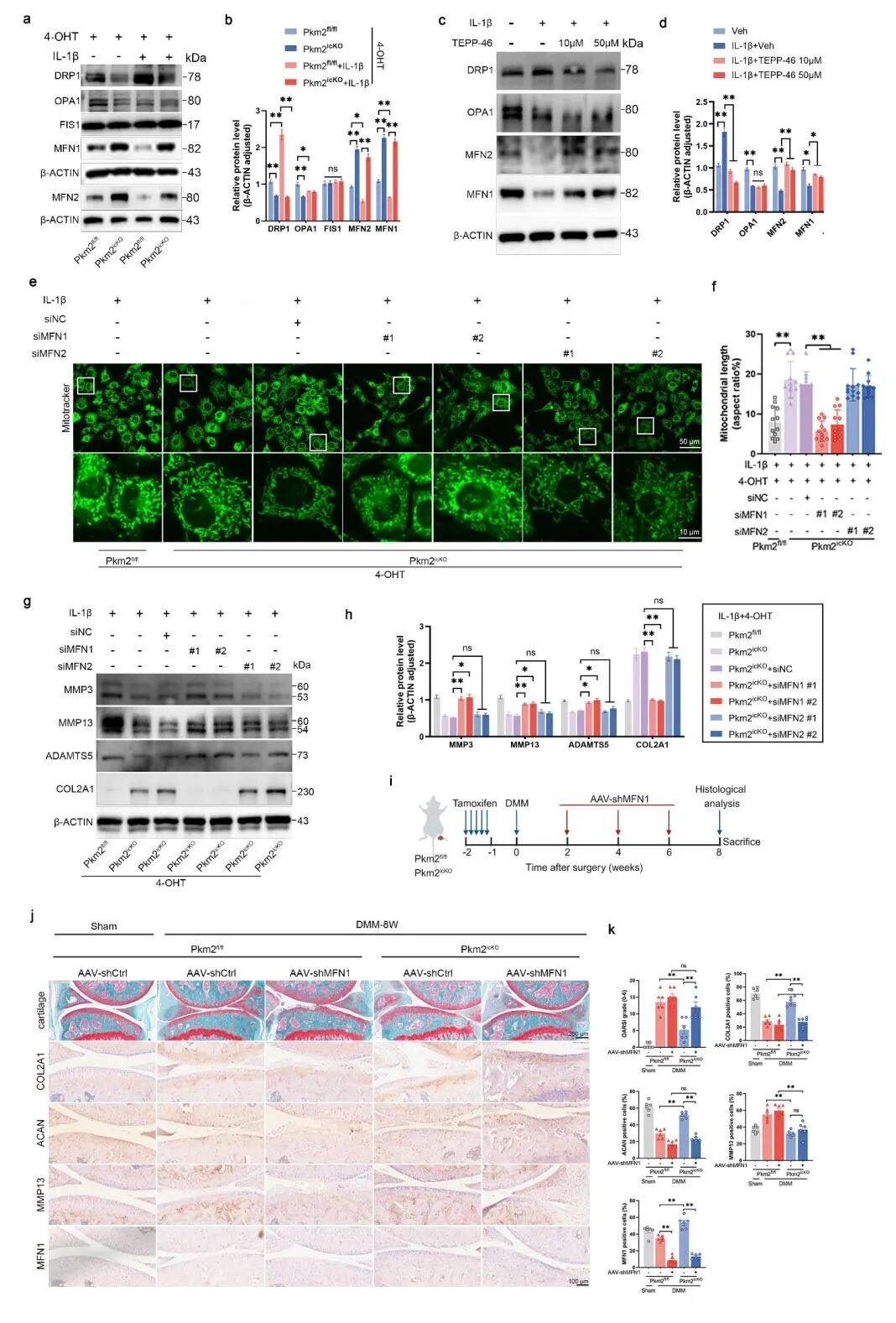
[A-D]:Pkm2 缺失或 TEPP-46 治疗均显著上调了 MFN1 和 MFN2 的表达,并抑制了 DRP1。
[E-F]:siRNA 干扰实验表明,只有敲低 Mfn1 (而非 Mfn2) 能消除 Pkm2icKO 带来的线粒体融合增强效应。
[G-H]:同样,敲低 Mfn1 能够阻断 Pkm2 缺失对基质代谢的保护作用,导致 COL2A1 下降及 MMPs 上升。
[I-K]:体内 AAV 介导的 shMFN1 注射实验证实,MFN1 的缺失完全抵消了 Pkm2icKO 的软骨保护表型。
发现点8:二聚体 PKM2 通过直接结合并激活 ERK 信号通路抑制 MFN1 的转录
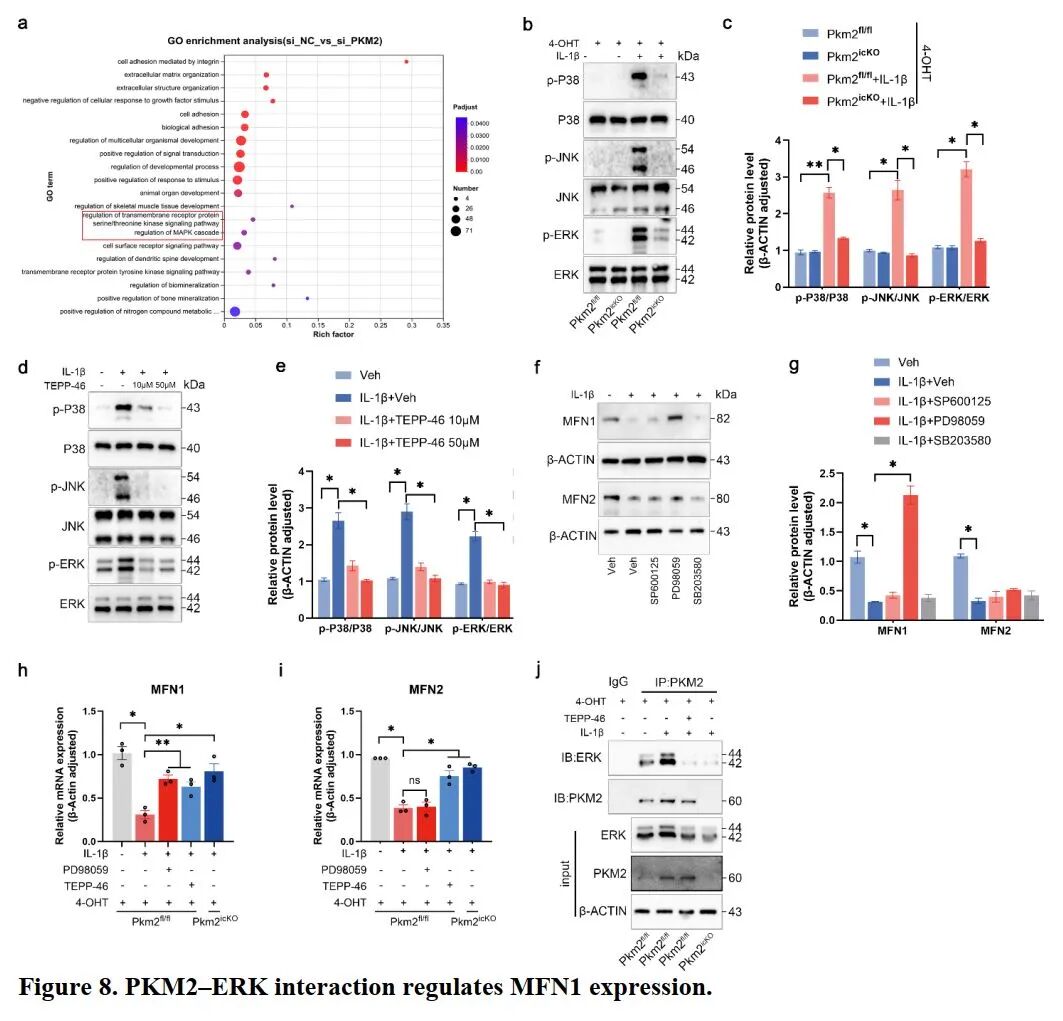
[A]:GO 分析显示 Pkm2 敲低显著影响了 MAPK 信号级联反应。
[B-E]:Pkm2 缺失或 TEPP-46 治疗均显著抑制了 IL-1β 诱导的 p38、JNK 及 ERK 的激活(磷酸化)。
[F-G]:使用抑制剂发现,只有 ERK 抑制剂 PD98059 能特异性恢复 MFN1 的蛋白水平,对 MFN2 无效。
[H-I]:mRNA 检测证实 MFN1 的转录受 ERK 信号通路精准调控。
[J]:内源性 Co-IP 实验证实 PKM2 与 ERK 存在直接物理相互作用,且二聚化状态进一步加强了这种结合。
Innovation & Takeaway
✨ 创新点
首次揭示了PKM2构象切换而非单纯表达水平对软骨细胞线粒体稳态的调控作用 ,并确定了MFN1是该途径的核心下游效应因子。
✨ 科研启示
本研究再次证明了“代谢重塑”不仅是肿瘤的特征,也是退行性疾病的核心。线粒体动态平衡(融合与分裂)受胞质代谢酶构象的精细调控,为OA机制研究提供了新视角。✨ 临床/应用价值
针对PKM2构象的调节剂(如TEPP-46)可能成为骨关节炎的一种新型非手术治疗手段,有望通过“稳态修复”而非单纯对症止痛来延缓软骨退变进程。
局限性
连接二聚体PKM2与通过ERK/MFN1轴的线粒体分裂/融合平衡的详细机制仅在DMM诱导的小鼠中建立,并且该调节轴未在人软骨细胞中进行验证,此外,尽管Pkm2icKO小鼠中MFN1和MFN2的蛋白水平均有所增加,但该研究仅侧重于MFN1,而未探索OA中MFN2下调的机制及其受PKM2潜在的调节,此外,信号传导的一些方面仍未阐明,例如,作为ERK抑制仅部分挽救MFN1表达,可能存在其他MAPK通路也参与该过程,最后,尽管TEPP-46是局部给予关节,但已知系统使用该药物具有代谢效应,且未彻底解决在非关节组织中的长期脱靶效应。
医脉通是专业的在线医生平台,“感知世界医学脉搏,助力中国临床决策”是平台的使命。医脉通旗下拥有「临床指南」「用药参考」「医学文献王」「医知源」「e研通」「e脉播」等系列产品,全面满足医学工作者临床决策、获取新知及提升科研效率等方面的需求。
