

医脉前沿 | MDS向AML转化机制新解:从克隆演化到微环境重塑

MDS进展为AML-MRC的生物学本质亟待厘清
骨髓增生异常综合征 (MDS)向继发性急性髓系白血病(sAML)的转化,在世界卫生组织(WHO)与2022年国际共识分类(ICC)中被定义为伴骨髓增生异常相关改变的急性髓系白血病(AML-MRC)。这一转化过程涉及细胞内在遗传事件与外在微环境重塑的动态相互作用,但直至近年,其完整的生物学图景仍不清晰。2020年Menssen与Walter曾系统综述MDS向继发性白血病进展的遗传学基础,并指出需借助RNA测序等先进技术深化认知。
(MDS)向继发性急性髓系白血病(sAML)的转化,在世界卫生组织(WHO)与2022年国际共识分类(ICC)中被定义为伴骨髓增生异常相关改变的急性髓系白血病(AML-MRC)。这一转化过程涉及细胞内在遗传事件与外在微环境重塑的动态相互作用,但直至近年,其完整的生物学图景仍不清晰。2020年Menssen与Walter曾系统综述MDS向继发性白血病进展的遗传学基础,并指出需借助RNA测序等先进技术深化认知。
近日,医脉通关注到一项发表于《Frontiers in Immunology》的前瞻性综述并进行解读,该研究系统梳理了MDS向AML转化过程中内在克隆演化与外在微环境重塑的双重机制,为临床风险分层与治疗策略优化提供了新视角。
核心机制:克隆演化与微环境重塑并行驱动MDS转化
MDS向AML转化,并非单一的基因突变累积过程,而是造血干/祖细胞(HSPCs)内在遗传改变与骨髓微环境(BMME)病理重塑协同作用的结果(图1)。
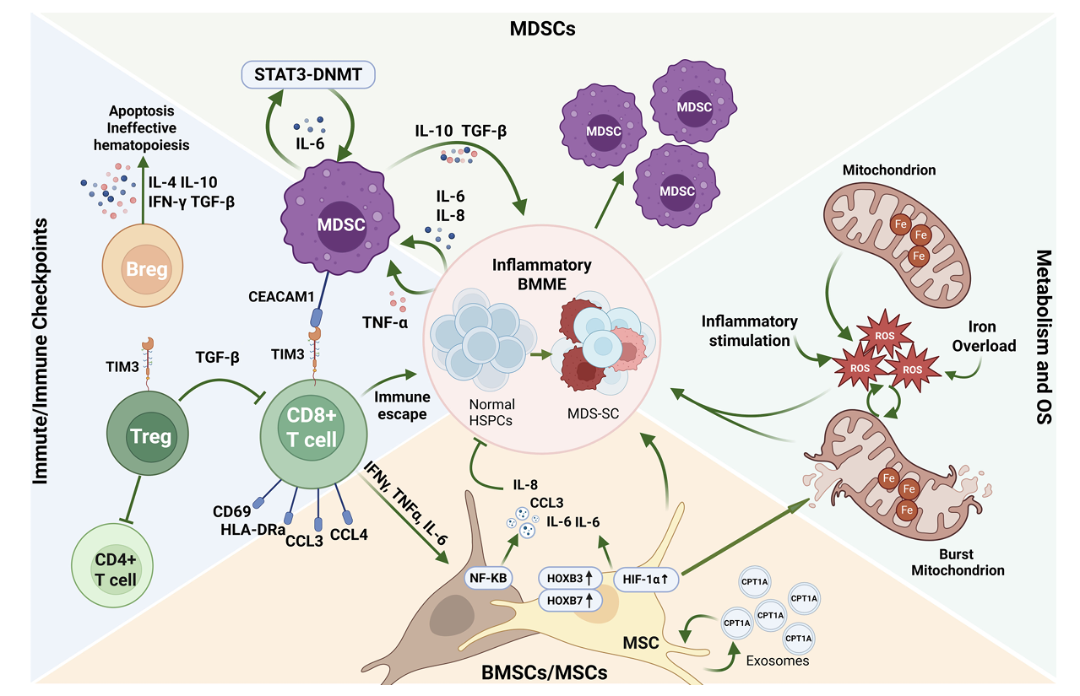
图1 从MDS到AML-MRC的并行克隆演化模型
克隆演化存在高度异质性
研究表明,MDS的进展几乎总是伴随着克隆演化。研究表明,MDS干细胞并非单一线性演化,而是呈现出多路径并存的复杂模式:
免疫表型异质性:MDS干细胞在免疫表型上可分为以常见髓系祖细胞(CMPs)或粒-单核系祖细胞(GMPs)为主的两种亚型,分别受TP53、RUNX1、DNMT3A等突变驱动。
克隆演化路径:单细胞DNA测序揭示了两类演化路径——“静态型”(以DNA甲基化基因突变为基础,累积突变负荷)与“动态型”(伴随新发染色体
异常或TP53、信号通路基因突变,克隆结构重塑)。染色质可及性改变:MDS干细胞在进展过程中逐渐失去干性染色质状态,过早经历髓系转录激活,提示表观调控异常亦是转化关键。
端粒生物学在克隆选择中的双重作用
端粒过短:与高风险MDS、低生存率相关,在端粒较短的情况下,干细胞的损耗加上衰老,会推动克隆演化。
端粒过长:如POT1突变或剪接因子突变(SRSF2、SF3B1等)所致,虽绕过复制衰老,却也促进克隆扩增。
治疗靶点:端粒酶
抑制剂imetelstat可抑制恶性干细胞,同时对正常细胞的影响较小。在改善患者红细胞生成方面已显示出显著的临床效果。
关键基因突变的功能解析
综述重点分析了多个与MDS进展密切相关的基因突变:
STAG2:约10%的MDS患者携带该突变,作为黏连蛋白复合体成分,其突变导致HSPCs积累、分化障碍,并与去甲基化药物(HMA)耐药相关。
RUNX1:约10%的MDS病例携带该突变,与-7/7q异常相关,突变后诱导DNA修复障碍与细胞衰老,推动疾病进展。GATA2:诱导造血破坏,作为儿童早期MDS的体细胞驱动突变。
GATA2:诱导造血破坏,作为儿童早期MDS的体细胞驱动突变。
TET2:TET2失活导致IL-6表达增加、NLRP3炎症小体激活和IL-1β产生,促进炎症性BMME和持续免疫抑制。
SRSF2/U2AF1:突变导致HSPCs中caspase 8异常剪接,破坏焦亡、凋亡和坏死性凋亡的调控。与ASXL1共突变可加速疾病进展,增加严重程度,促进向AML-MRC转化。
TP53:TP53是位于17p染色体上的肿瘤抑制基因,编码转录因子p53。突变通常与复杂核型相关,预后极差。
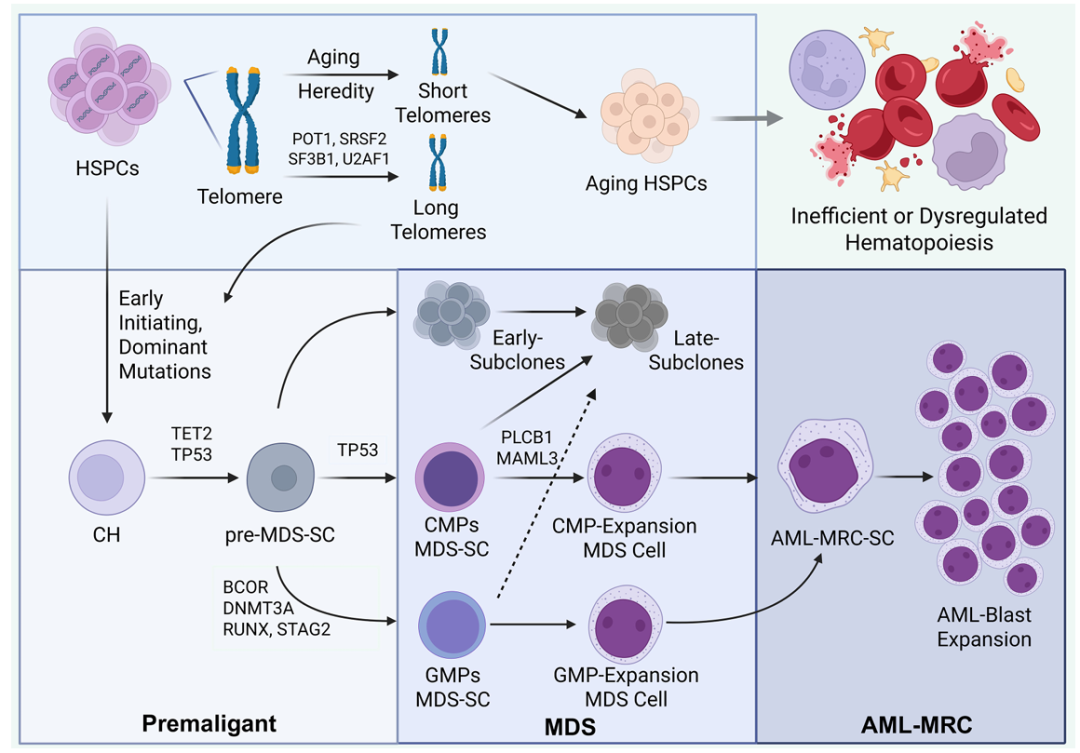
图2 端粒生理学和克隆进化过程的复杂性
在MDS进展过程中,BMME从支持正常造血转向促进恶性克隆扩张。
骨髓基质细胞(BMSCs):在LR-MDS中出现炎症重编程,由浸润的CD8+ T细胞驱动,表现出高水平的激活标志物,其高表达TNFRSF9、CD69、HLA-DRA等激活标志物,并分泌TNF-α、IFN-γ、CCL3和CCL4等细胞因子。关键的是,这种炎症转变独立于造血克隆中的特定遗传驱动因素,通过赋予突变HSPCs相对生存优势促进克隆演化。
间充质干细胞(MSCs):MDS-MSCs表现出衰老、炎症、代谢异常,丧失对正常造血的支持能力,反而促进MDS细胞存活。其外泌体中CPT1A富集,破坏自身代谢稳态。
髓系抑制细胞(MDSCs):在MDS-BMME中扩增,表现出强免疫抑制表型,特征为IL-10和TGF-β1分泌升高。其关键的自我强化生存机制涉及自分泌IL-6驱动的STAT3-DNMT表观遗传轴,该轴沉默TNF-α表达,促进其持续积累,并营造对PD-1靶向治疗耐药的微环境。
免疫网络:MDSCs丰度与Tregs增加呈正相关,共同抑制细胞毒性T细胞和自然杀伤(NK)细胞功能,实现恶性克隆的免疫逃逸。树突状细胞和单核细胞功能同样受损,显示启动有效抗肿瘤T细胞反应的能力降低。TIM3是MDS免疫失调的核心调节因子。在恶性干细胞上,TIM3表达标志着一个独特的生物学群体:尽管形态上与TIM3-细胞相似,但TIM3+ MDS干细胞表现出更低的集落形成能力、更多核型异常,以及分化阻滞伴增殖和凋亡抗性增加。在免疫区室,TIM3在MDS的Treg细胞上高表达,与TGF-β分泌相关。阻断TIM3和CEACAM1可部分逆转CD8+ T细胞耗竭,代表一种潜在的治疗策略,Sabatolimab等单克隆抗体正在临床研究中。
能量代谢异常与氧化应激:线粒体分裂异常、铁过载、HIF-1α/mTOR通路激活共同导致活性氧(ROS)升高,推动DNA损伤与克隆演化。mTOR信号上调DNMT1表达,将代谢异常与表观遗传调控直接联系。
临床挑战:HMA耐药与移植困境——MDS向AML转化的治疗瓶颈
HMA(阿扎胞苷、地西他滨)是高危MDS(HR-MDS)患者无法接受造血干细胞移植(HSCT)时的标准治疗方案。然而,HMA的总体反应率不足50%,且一旦出现耐药,患者的中位总生存期(mOS)通常不足6个月。HMA耐药后接受免疫治疗的获益有限,现有挽救性治疗方案难以突破耐药瓶颈。
研究显示,HMA治疗过程中,PD-1表达水平在治疗前4个周期内显著上调,且治疗中高表达PD-1可独立预测后续AML转化和生存期缩短。提示HMA虽可靶向克隆,但也可能诱导T细胞耗竭表型,促成免疫逃逸。一项基于CRISPR-Cas9筛选的研究发现,E3泛素连接酶TOPORS缺失可增强白血病干细胞对HMA的敏感性。TOPORS缺失导致DNA损伤应答受损,SUMO化DNMT1积累,从而增强HMA的细胞毒作用。这一通路为逆转HMA耐药提供了潜在靶点。
Venetoclax联合HMA在特定基因型(如U2AF1突变)患者中显示出良好疗效,而TP53突变患者即使接受移植,预后仍差,提示需探索新型移植后维持策略。
MDS向AML的转化是一个多因素、多阶段的动态过程,涉及HSPCs内在演化与BMME外在重塑的双向互动。未来风险分层模型需整合免疫、代谢、遗传等多组学数据,治疗策略也应从单一靶向克隆,转向同时干预微环境与代谢依赖。随着单细胞多组学技术的应用,MDS进展的全景图正逐步清晰。医脉通将持续关注这一领域的最新进展,为临床医生提供前沿、实用的学术解读。
参考文献:
Zhang Y, et al. Myelodysplastic syndrome progress to acute myeloid leukemia: new insights and updates. Front Immunol. 2026 Feb 4;17:1769944.
编辑:Felicia
审校:Janet
排版:Wyn
执行:Baa
,助力中国临床决策”是平台的使命。医脉通旗下拥有「临床指南」「用药参考」「医学文献王」「医知源」「e研通」「e脉播」等系列产品,全面满足医学工作者临床决策、获取新知及提升科研效率等方面的需求。声明:本平台旨在为医疗卫生专业人士传递更多医学信息。本平台发布的内容,不能以任何方式取代专业的医疗指导,也不应被视为诊疗建议。如该等信息被用于了解医学信息以外的目的,本平台不承担相关责任。本平台对发布的内容,并不代表同意其描述和观点。若涉及版权问题,烦请权利人与我们联系,我们将尽快处理。

